ATAC-seq - A Tool for Analyzing Epigenetics
ATAC-seq - A Tool for Analyzing Epigenetics
Blog Article
Chromatin Dynamics and Gene Regulation in Eukaryotic Cells
The total length of DNA within human cells approximates two meters and necessitates complex folding processes to be accommodated within the nucleus. During this dynamic folding process, chromatin exhibits heterogeneous structural states, manifesting as either open or closed configurations. The open state of chromatin is crucial for the regulation of gene expression: transcription predominantly occurs in regions where chromatin is more accessible. Additionally, DNA methylation, histone modifications, and the binding of transcription factors require access to these open chromatin regions.
Open chromatin, characterized by a lower degree of compaction, provides a platform for the action of transcriptional regulatory proteins, forming the foundation for gene transcription and epigenetic regulation. The degree of chromatin folding and the presence of open regions are focal points of epigenomic research. Studies on chromatin accessibility can elucidate the molecular mechanisms of gene regulation and epigenetic changes, revealing the interaction networks between various transcriptional regulatory factors and downstream gene expression from a structural perspective.
In eukaryotic cells, chromatin serves as the fundamental unit of heredity, composed of DNA and histone proteins, and regulates cell-specific gene expression (Jackson Dean A., 1995; Bártová Eva and Kozubek Stanislav, 2012). As a dynamic nuclear structure, chromatin exhibits transcriptional activity during interphase and relative inactivity during metaphase of the cell cycle (Pederson Thoru, 2004). Transcriptional regulation involves the dynamic interactions between chromatin structure and numerous transcription factors recruited to enhancers, upstream activator sequences, and proximal promoter elements. These transcription factors facilitate the recruitment of RNA polymerase to core promoters, initiating transcription (Gottesfeld Joel M. and Carey Michael F., 2018).
Generally, regulatory elements selectively localize to accessible chromatin, which is essential for transcriptional regulation (Thurman et al., 2012). Although the occupancy of transcription factors is not always positively correlated with chromatin accessibility (Hsiung et al., 2015), maintaining an accessible chromatin conformation is necessary for the activation of target genes through transcription factor binding (Morris et al., 2014). Conversely, heterochromatin (or closed chromatin) restricts the binding of transcription factors and regulatory elements to promoters or enhancers, leading to gene silencing (Stergachis et al., 2013; Rinn J L and Chang H Y., 2012; Chen T and Dent S Y R, 2014). In summary, gene transcription necessitates the unwinding of higher-order DNA structures, though not entirely; only the regions of genes to be expressed need to be opened. This process is primarily facilitated by chromatin histone modifications, particularly acetylation. These partially opened chromatin regions are referred to as open chromatin, and once opened, they permit the binding of regulatory proteins, such as transcription factors.
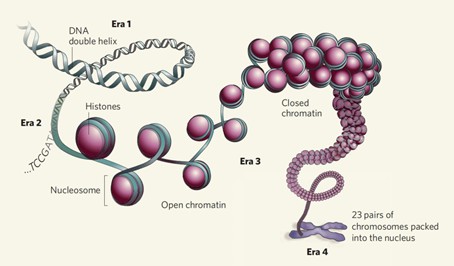
Figure 1 Open Chromatin vs Closed Chromatin
Chromatin Accessibility and Its Implications for Transcriptional Regulation
Chromatin accessibility refers to the degree of openness of chromatin, primarily facilitated by histone modifications. This property indicates the extent to which regulatory factors can bind to open chromatin regions and is closely associated with transcriptional regulation. Investigating regions of open chromatin in specific cellular states provides insights into transcriptional regulatory mechanisms at the DNA level.
High-Throughput Methodologies in Epigenetic Research for Studying Chromatin Accessibility
Research in epigenetics predominantly employs high-throughput, genome-wide methodologies to study chromatin accessibility. Traditional experimental approaches, including Micrococcal MNase-seq and DNase-seq, operate on the principle that chromatin becomes more accessible when its structure is relaxed, resulting in decreased aggregation of DNA and histones. This exposure of DNA allows it to be cleaved by nucleases such as MNase or DNase I. Sequencing of the cleaved DNA fragments, followed by comparison with known genomic sequences, enables the identification of regions with increased accessibility. However, these methods are often labor-intensive and exhibit low reproducibility.
Another technique, FAIRE-seq, involves ultrasonic lysis followed by phenol-chloroform extraction, circumventing the need for nucleases or antibodies. Despite this advantage, FAIRE-seq suffers from high background noise, low signal-to-noise ratios, and challenges in optimizing formaldehyde crosslinking times.
ChIP-seq is employed to identify the binding sites of specific transcription factors or protein complexes, thereby elucidating DNA-protein interactions. This method utilizes antibodies to enrich for protein-DNA complexes, followed by sequencing of the associated DNA.
Although these techniques share a common analytical framework of identifying enriched regions and performing functional analyses, each has distinct limitations. In comparison, Assay for ATAC-seq utilizes the Tn5 transposase and is advantageous due to its simplicity, high reproducibility, and minimal requirement for cell or tissue samples. Additionally, ATAC-seq generates highly robust signals, making it the preferred technique for studying chromatin accessibility.
The Principle and Process of ATAC-seq
ATAC-seq, or Assay for Transposase-Accessible Chromatin with high-throughput sequencing, is a method developed in 2013 by the laboratories of William J. Greenleaf and Howard Y. Chang at Stanford University for investigating chromatin accessibility (often referred to as chromatin openness).
ATAC-seq represents an innovative technique in epigenetic research, leveraging the highly active Tn5 transposase as a probe to cleave DNA sequences and thereby locate accessible regions of chromatin across the entire genome (Buenrostro et al., 2013). DNA transposition involves the relocation of DNA transposons from one region of the chromosome to another with the assistance of transposase enzymes (Chuong et al., 2017). For ATAC-seq, this process necessitates that the insertion sites be in open chromatin regions. The methodology involves the artificial introduction of transposases carrying known DNA sequence tags into the cell nucleus, where they cleave the open chromatin, yielding DNA fragments tagged with primer sequences. These tagged fragments are then amplified using the known sequence primers to construct sequencing libraries (Buenrostro et al., 2013).
The most commonly used transposase in this context is the Tn5 transposase, which preferentially inserts into accessible chromatin regions over inaccessible ones. Functioning as a probe, the Tn5 transposase employs a "cut-and-paste" mechanism to detect chromatin accessibility on a genome-wide scale. The resulting DNA fragments are marked with sequencing tags to identify unprotected regions of the DNA (Reznikoff William S., 1993; Haniford D.B. and Ellis M.J., 2015).
In simpler terms, the Tn5 transposase can randomly bind to and cleave DNA in open chromatin regions while simultaneously inserting adaptor sequences at the cleavage sites. By incubating the transposase complex, which includes red and green sequence tags, with the cell nuclei and subsequently performing PCR amplification using the known sequence tags, a library is formed. Sequencing this library reveals information about the open chromatin regions.
The construction of an ATAC-seq library involves three main steps: nuclei preparation, transposition, and amplification. Initially, the tissue or cells to be examined are suspended to form a homogeneous single-cell suspension, followed by incubation in a lysis buffer to generate crude nuclei (Figure 3A). Subsequently, the suspended nuclei are incubated in a transposition reaction mixture, producing DNA fragments (Figure 3B). Finally, the transposed DNA is amplified to generate a sequencing library (Figure 3C). The reaction between the transposase and the sample chromatin is a critical step in the ATAC-seq experiment (Picelli et al., 2014).
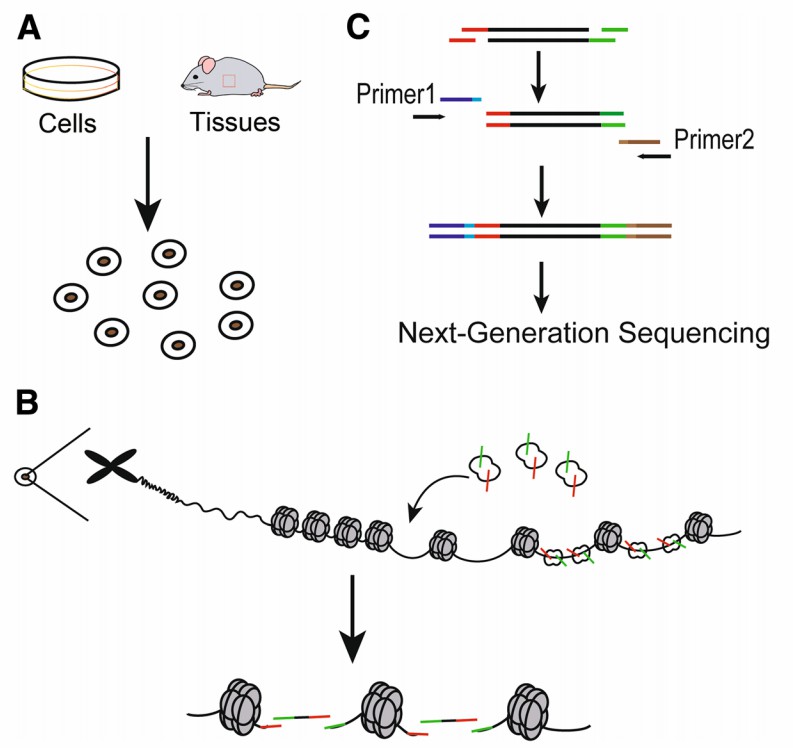
Figure 2 illustrates the main steps of ATAC-seq (Sun et al., 2019). (A) Nuclei Preparation: Target cells are lysed in lysis buffer to collect nuclei. (B) Transposase Reaction: Genomic DNA is tagged with Tn5 transposase. The green symbols represent "sequencing adapter 1" of the Tn5 transposase, while the red symbols represent "sequencing adapter 2". (C) PCR Amplification: Sequencing libraries are generated using PCR primers 1 and 2. Primers 1 and 2 are two universal PCR primers that capture specific length fragments and add barcodes suitable for next-generation sequencing.
Note: Although the experimental process is illustrated using animal experiments, it is equally applicable to plants.
Quality Control and Data Analysis in ATAC-seq
Prior to sequencing, ATAC-seq libraries require rigorous quality control to ensure that the library concentration meets sequencing standards. Sequencing is performed on libraries that meet these criteria, and raw reads are collected. Quality assessment and data filtering are subsequently conducted to obtain clean reads (Davie et al., 2015; Bell et al., 2011; Mardis, 2008). Post-filtering, high-quality reads approximately 150 nucleotides (nts) in length are processed for further analysis (Miskimen et al., 2017).
Peak calling involves mapping reads to the reference genome to identify accessible chromatin regions, such as promoters, enhancers, and insulators (Kumasaka et al., 2016; Ackermann et al., 2016; Quillien et al., 2017). This allows for a series of detailed analyses, including determining the genome-wide distribution of reads, assessing peak length distribution, performing functional analysis of genes associated with identified peaks, examining the distribution of peaks within gene functional elements, and analyzing differential peaks between samples (Pranzatelli et al., 2018; Mu et al., 2012).
Advantages and Limitations of ATAC-seq Technology
Comparison of ATAC-seq with Other Techniques
ATAC-seq, or Assay for Transposase-Accessible Chromatin with high-throughput sequencing, was initially developed as an alternative or complementary method to MNase-seq, FAIRE-seq, and DNase-seq (Table 1).
In MNase-seq and DNase-seq, the reduction in DNA and histone aggregation leads to the exposure of unprotected DNA, which can then be cleaved by nucleases such as MNase and DNase. By sequencing these cleaved DNA fragments and comparing them to a reference genome, regions of accessible chromatin can be identified. However, these methods are often time-consuming and exhibit poor reproducibility. FAIRE-seq involves the fixation of DNA with formaldehyde, followed by phenol-chloroform extraction to isolate exposed DNA. Despite its utility, FAIRE-seq suffers from high background noise, low signal-to-noise ratio, and challenges in optimizing formaldehyde crosslinking times.
ATAC-seq addresses several of these limitations by employing the Tn5 transposase to insert sequencing adapters into open chromatin regions, thereby allowing for the efficient identification of accessible chromatin with minimal starting material. This method is advantageous due to its simplicity, rapid execution, and high reproducibility, making it a preferred technique for chromatin accessibility studies.
Table 1 Comparison of several sequencing methods (Sun et al., 2019).
| Method | MNase-seq | DNase-seq | FAIRE-seq | ATAC-seq |
| Cell State | Any | Any | Any | Fresh cells or slowly cooled frozen cells |
| Principle | MNase digestion of DNA protected by proteins or chromatin-bound nucleosomes. | Preferential cleavage of DNA sequences lacking nucleosomes by DNaseI. | Separation of naked DNA based on formaldehyde fixation and phenol-chloroform extraction. | Insertion of Tn5 transposase into DNA sequences not protected by proteins or nucleosomes and excising them. |
| Target Region | Focuses on nucleosome positioning. | Accessible chromatin regions, concentrated at transcription factor binding sites. | Accessible chromatin regions. | Genome-wide accessible chromatin regions, including transcription factors and histone modifications. |
| Specific Features | 1. Large number of cells as starting material; 2. Accurate enzyme quantities required; 3. Localization of entire nucleosomes and inactive regulatory regions; 4. Detection of inactive regions by degrading active ones; 5. Standard analysis requires 150-200M reads. | 1. Large number of cells as starting material; 2. Complex sample preparation process; 3. Accurate enzyme quantities required; 4. Standard analysis requires 20-50M reads. | 1. Low signal-to-noise ratio complicates data analysis; 2. Results heavily reliant on formaldehyde fixation; 3. Standard analysis requires 20-50M reads. | 1. Less starting material required; 2. Standard analysis requires 20-50M reads by reducing sequencing depth; 3. Convenient access to genome-wide accessible chromatin regions; 4. Impact of mitochondrial data on result accuracy. |
Advantages of ATAC-seq Technology
Efficiency and Time Reduction: The utilization of a transposase-based approach markedly reduces the experimental duration to approximately 2-3 hours. This expedited timeline is achieved through a direct enzymatic reaction for DNA fragmentation, obviating the labor-intensive conventional procedures of DNA shearing, end-repair, and adapter ligation.
Simplified Protocol: The streamlined experimental workflow minimizes sample preparation duration and diminishes the likelihood of errors, consequently enhancing the success rate and reproducibility of experiments. In contrast, DNase-seq and MNase-seq protocols typically entail 2-3 days, while FAIRE-seq necessitates 3-4 days.
Reduced Sample Requirement: The prerequisite sample input is substantially reduced by at least 1000-fold, from 1 million cells (FAIRE-seq) and 50 million cells (DNase-seq) to approximately 500 cells. This reduction is particularly advantageous in scenarios where sample collection poses challenges.
High-Resolution Mapping: ATAC-seq leverages paired-end sequencing to generate intricate nucleosome positioning and occupancy maps. Paired-end sequencing enables the sequencing of both ends of DNA fragments, thereby facilitating precise alignment of reads to repetitive genomic regions.
Enhanced Reproducibility and Simplicity: The reproducibility of ATAC-seq surpasses that of MNase-seq and DNase-seq. Moreover, the methodology is characterized by its simplicity of execution, minimal sample input requirements, and production of superior-quality sequencing signals.
Single-Cell Sequencing Capability: Recent advancements underscore the significance of single-cell sequencing in personalized epigenetic investigations. Conventional techniques such as ChIP-seq, DNase-seq, and MNase-seq are incompatible with single-cell analyses. However, ATAC-seq has been experimentally validated for single-cell sequencing, rendering it a cornerstone in contemporary epigenetic research.
Limitations of ATAC-seq Technology
Random Sequencing Adapters: The Tn5 transposase employs a "cut-and-paste" mechanism, fragmenting and tagging DNA in unprotected regions with sequencing adapters. Each DNA fragment's sequencing adapters at both ends are random, resulting in a 50% chance of both ends having identical sequencing adapters. Consequently, approximately half of the generated fragments are unusable for subsequent enrichment, amplification, and sequencing.
Preference for Unbound DNA and Transcription Factor Binding Sites: Studies indicate that "naked" DNA devoid of nucleosomes and transcription factors is more susceptible to cleavage by Tn5 transposase. Additionally, Tn5 transposase tends to bind and cleave in transcription factor binding regions, leading to partial loss of transcription factor information. These limitations render ATAC-seq challenging for detecting transcription factor footprints, which are crucial for identifying potential binding motifs.
Inclusion of Mitochondrial Reads: Due to the presence of mitochondrial DNA, data obtained through ATAC-seq inevitably contain some mitochondrial reads. Depending on the cell type, ATAC-seq data may comprise 20-80% mitochondrial sequencing reads.
Differences Between ATAC-seq and ChIP-seq
ChIP-seq and assay for transposase-accessible chromatin using sequencing (ATAC-seq) are distinct methodologies employed in the study of chromatin dynamics and transcription factor binding.
ChIP-seq: This technique is predicated on the pre-identification of a specific transcription factor of interest. An antibody targeting the specific transcription factor is utilized to precipitate the DNA bound to it during the ChIP process. This enables the subsequent sequencing of the co-precipitated DNA, thereby validating the interaction between the transcription factor and the DNA.
ATAC-seq: In contrast, ATAC-seq does not focus on any specific transcription factor. Instead, it assesses the overall chromatin accessibility across the entire genome. This technique identifies potential binding sites for various proteins based on the openness of chromatin regions. By combining ATAC-seq with other methodologies, researchers can screen for and identify regulatory elements and factors of interest on a genome-wide scale.
In summary, while ChIP-seq provides insights into the binding interactions of specific transcription factors with DNA, ATAC-seq offers a broader perspective on chromatin accessibility and potential protein binding sites across the genome.
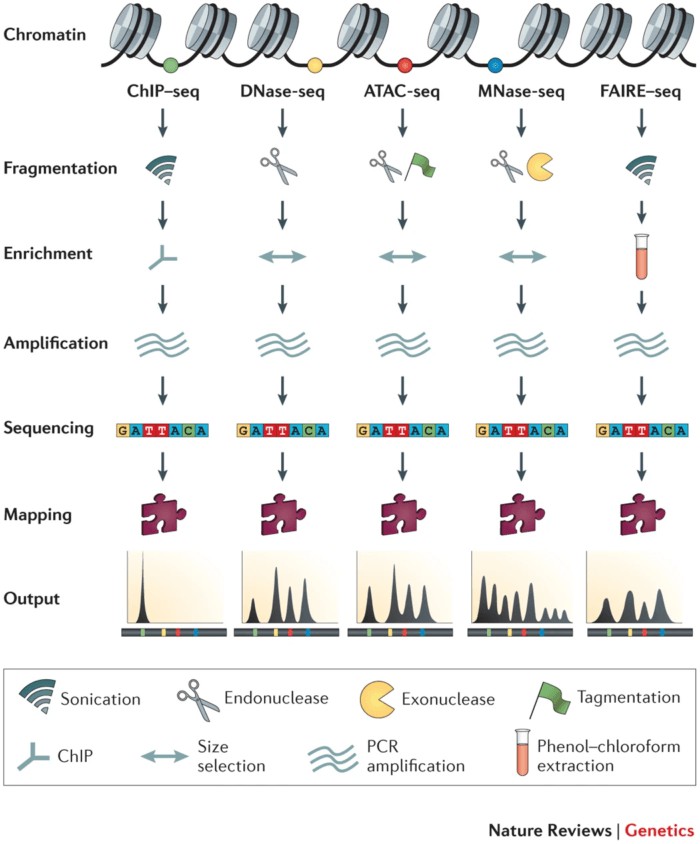
Figure 3 High-Throughput Methodologies Studying Chromatin Accessibility
Applications of ATAC-seq
Nucleosome Positioning
The Assay for ATAC-seq is extensively employed for the delineation of nucleosome positions across the genome, thereby providing significant insights into chromatin organization. Buenrostro et al. (2013) demonstrated the application of ATAC-seq in profiling nucleosome occupancy and chromatin accessibility in human cell lines, elucidating patterns of nucleosome positioning associated with regulatory elements. This study underscored the efficacy of ATAC-seq in generating high-resolution nucleosome positioning maps, which are essential for understanding the regulatory landscape of the genome.
Identification of Key Transcription Factors
The analysis of open chromatin regions via Assay for ATAC-seq facilitates the identification of transcription factors implicated in gene regulation. Corces et al. (2016) utilized ATAC-seq to profile chromatin accessibility across diverse human tissues, identifying transcription factors such as CTCF (CCCTC-binding factor) and AP-1 (Activator Protein 1) as crucial regulators of gene expression. This investigation highlighted the importance of ATAC-seq in elucidating transcription factors that play essential roles in cell-specific regulatory networks.
Identification of Promoter Regions, Potential Enhancers, or Silencers
ATAC-seq is adept at identifying open chromatin regions that correspond to promoters and distal regulatory elements, encompassing enhancers and silencers. Thurman et al. (2012) employed ATAC-seq to map DNase I hypersensitive sites within the human genome, thereby revealing active regulatory elements such as promoters and enhancers. This methodology is instrumental in elucidating the regulatory elements that drive gene expression and in understanding the complex interactions within the genome.
Integration with Multi-omics
The integrative approach of combining Assay for ATAC-seq with other omics technologies, such as RNA sequencing, provides a comprehensive perspective on gene regulation. For instance, Satpathy et al. (2019) employed a combination of ATAC-seq and single-cell RNA-seq to investigate the immune cell landscape in melanoma, thereby identifying regulatory elements associated with gene expression changes across different cell states. This integrative methodology enhances the understanding of how chromatin accessibility influences gene expression and cellular function.
Chromatin Accessibility Mapping
Assay for ATAC-seq generates comprehensive genome-wide maps of chromatin accessibility, imperative for investigating dynamic changes in chromatin structure. Schep et al. (2015) utilized ATAC-seq to map chromatin accessibility during mouse embryonic stem cell differentiation, revealing dynamic alterations correlating with gene expression patterns. These maps are crucial for elucidating the epigenetic regulation of developmental processes.
Identification of Target Genes
Regulated by Transcription Factors, Assay for ATAC-seq aids in identifying target genes controlled by transcription factors by locating regions of open chromatin where these factors bind. Heinz et al. (2018) employed ATAC-seq to pinpoint binding sites of transcription factors such as PU.1 and CEBPA in macrophages, elucidating their roles in regulating immune response genes. This application is vital for mapping the regulatory networks that govern gene expression in diverse biological contexts.
Characterization of Transcription Factor Binding Sites (Footprinting)
ATAC-seq can be utilized for the characterization of transcription factor binding sites through footprinting analysis. Pique-Regi et al. (2011) applied ATAC-seq footprinting to identify protected regions within open chromatin, indicative of direct binding sites for transcription factors such as NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) and p53. This technique provides detailed insights into the binding dynamics of transcription factors and elucidates their regulatory roles.
Case Study
Case 1
Title: ATAC-seq and RNA-seq reveal the role of AGL18 in regulating fruit ripening via ethylene-auxin crosstalk in papaya
Methods: ATAC-seq, RNA-seq, Subcellular Localization, Y1H, Dual Luciferase Reporter Gene Assay, ChIP-qPCR, EMSA
Abstract:
This study employs integrated ATAC-seq and RNA-seq to delineate chromatin accessibility patterns under different 1-methylcyclopropene (1-MCP) treatments, uncovering key transcription factors and target genes influencing fruit ripening. The approach is straightforward yet informative, conducive to producing publications of approximately five points.
Background:
Papaya (Carica papaya) stands as a quintessential tropical fruit, prized for its distinctive flavor and rich nutritional profile. However, papaya is a climacteric fruit, undergoing rapid physiological changes during ripening, rendering it highly susceptible to rot, thereby posing significant challenges for its transportation and storage (Sivakumar et al., 2013). Studies have shown that 1-methylcyclopropene (1-MCP), an ethylene receptor inhibitor, can delay ripening and rotting in fruits such as bananas, apples, and papayas, with appropriate 1-MCP treatment postponing papaya fruit yellowing and softening. Nonetheless, improper 1-MCP treatment can impede papaya ripening, resulting in fruits that fail to soften adequately and retain high hardness even at the end of storage, severely limiting the application of 1-MCP in postharvest papaya industries. Various researches indicate the pivotal roles of auxin and ethylene in fruit ripening. However, the molecular mechanisms underlying the interaction between auxin and ethylene during papaya fruit ripening remain elusive (Petrasek et al., 2009; Tian et al., 2006).
Researchers employed integrated ATAC-seq and RNA-seq analyses to unveil differences in chromatin accessibility under 1-MCP treatments of varying durations during papaya ripening, aiming to identify key differentially accessible regions (DARs), upstream motifs, and transcription factors (TFs) implicated in delaying and inducing abnormal papaya ripening under 1-MCP treatment. This elucidation aids in identifying genes that modulate the interplay between the ethylene and auxin signaling pathways associated with papaya ripening, thereby serving as candidate genes for improving fruit quality and postharvest shelf life (Cai et al., 2022).
Experimental Design:
Fruits were divided into three groups: short-term treatment with 400 nL/L 1-MCP for 2 hours, long-term treatment with 400 nL/L 1-MCP for 16 hours, and untreated control continuously kept in a sealed foam box for 16 hours. Samples were collected one and six days after treatment, with three biological replicates for RNA-seq and two biological replicates for ATAC-seq (Figure 4).
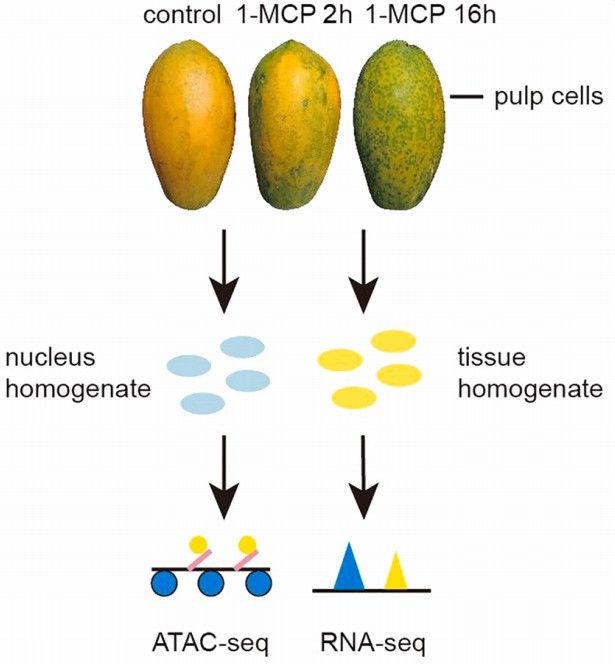
Figure 4 illustrates the workflow of ATAC-seq and RNA-seq on papaya fruits post 1-MCP treatment. Blue ellipses represent nuclear homogenate, while yellow ellipses represent tissue homogenate.
Report this page